The ei data frame holds the sample ID and condition information, but we need to combine this information with the cluster IDs. Again, save the counts table without header, we will need it later. ; Jiang, Y.M. RNA-sequencing is a powerful technique that can assess differences in global gene expression between groups of samples. ; Wang, Y.-S.; Gao, Y.-H.; Zhang, R.; et al. Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. This plot is a good check to make sure that we are interpreting our fold change values correctly, as well. This transcriptome is given to Salmon in the form of a (possibly compressed) multi-FASTA file, with each entry providing the sequence of a transcript1. Here we present the DEseq2 vignette it wwas composed using STAR and HTseqcount and then Deseq2. We use cookies on our website to ensure you get the best experience. In particular, many of the data wrangling steps were derived from this tutorial. For questions or other comments, please contact me. ; Liu, H.; Feng, X.-D.; Ma, D.-Y. ; Li, J.; Fang, J.P.; Liu, T.T. ; Jacobs, A. Next, we can get an idea of the metadata that we have for every cell. Previously, we performed QC on the Golden Snidget RNA sequencing data, aligned the sequencing reads to its genome, and obtained expression counts. ; Chen, M.L. RNA-Seq-DGE.rmd used to create output of the script shown in the PDF file here. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's You can RNA-seq workflow: gene-level exploratory analysis and differential expression: Here we are some examples of working on R on Counts. This type of RNAseq is as much of an art as well as science because WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. Wan, L.R. A Conserved Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W. In lessons 9 through 17 we will learn how to analyze RNA sequencing data. ; Landolin, J.M. The course is designed for PhD students and will be given at the University of Mnster from 10th to 21st of October 2016. Stanley-Samuelson, D.W.; Jurenka, R.A.; Cripps, C.; Blomquist, G.J. NOTE: The DESeq2 vignette suggests large datasets (100s of samples) to use the variance-stabilizing transformation (vst) instead of rlog for transformation of the counts, since the rlog function might take too long to run and the vst() function is faster with similar properties to rlog. If we treat cells as samples, then we are not truly investigating variation across a population, but variation among an individual. For instructions on importing for use with edgeR or limma, see the ; Yang, J.; Luo, R.; Tian, H.X. And the BORED and EXCITED groups do cluster together. HHS Vulnerability Disclosure, Bioinformatics Training and Education Program, Lesson 1: Introduction to Unix and the Shell, Lesson 2: Navigating file systems with Unix, Lesson 7: Downloading the RNA-Seq Data and Dataset Overview, Lesson 9: Reference genomes and genome annotations used in RNA sequencing, Lesson 10: Introducing the FASTQ file and assessing sequencing data quality, Lesson 11: Merging FASTQ quality reports and data cleanup, Lesson 13: Aligning raw sequences to reference genome, Lesson 15: Finding differentially expressed genes, Lesson 16: Classification based RNA sequencing analysis, Gene ontology and pathway analysis: PowerPoint slides, Database for Annotation, Visualization and Integrated Discovery (DAVID) - an overview, Introduction to Qiagen Ingenuity Pathway Analysis, Create a folder to store the Golden Snidget differential expression analysis results, Format the Golden Snidget counts table for differential expression analysis, Database for Annotation, Visualization and Integrated Discovery (DAVID) - practicing what we learned, U.S. Department of Health and Human Services. Biological invasion of European tomato crops by, Guimapi, R.A.; Srinivasan, R.; Tonnang, H.E. https://doi.org/10.3390/insects14040363, Liu, Min, Feng Xiao, Jiayun Zhu, Di Fu, Zonglin Wang, and Rong Xiao. Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. In Galaxy, download the count matrix you generated in the last section using the disk icon. WebTUTORIALS. After realignment with the NCBI for Biotechnology Information database, 21 differentially expressed cytochrome P450 genes were screened. WebI know DESeq2 was initially used for RNA-seq to detect the regulation of gene expressions. A new mathematical model for relative quantification in real-time RT-PCR. I am working with gene expression data from a RNASeq dataset using DESEq2. ; ; ; ; ; Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. ; Cao, Y.; Tian, L.; et al. ; Aguiar-Santana, I.A. We can use the functions from the SingleCellExperiment package to extract the different components. ; Wang, J.J. Genome-wide identification of long non-coding RNAs (lncRNAs) associated with malathion resistance in, Qiao, H.L. The -i argument tells salmon where to find the index -l A tells salmon that it should automatically determine the library type of the sequencing reads (e.g. One aliquot of PBMCs was activated by 100 U/mL of recombinant IFN- for 6 hours. ; Epton, M.J.; Gong, P.; Jin, L.; Condon, G.C. The -1 and -2 arguments tell salmon where to find the left and right reads for this sample (notice, salmon will accept gzipped FASTQ files directly). The color blocks indicate substructure in the data, and you would expect to see your replicates cluster together as a block for each sample group. The rest of the tutorial below will assume that youve placed the salmon executable in your path, so that simply running salmon will invoke the program. Full-length non-chimeric reads (FLNC) were clustered at the isoform level, and full-length transcripts were corrected using Proovread software and Illumina RNA-seq data to improve sequence accuracy. WebDESeq2 Tutorial This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. ; Ossa, G.A. Home; Blog; rnaseq deseq2 tutorial; rnaseq deseq2 tutorial. ; Brooks, A.N. Normalise to a housekeeping gene in DESEq2. permission is required to reuse all or part of the article published by MDPI, including figures and tables. A newly discovered invasive pest in China-, Guedes, R.N.C. The Basics of DESeq2 A Powerful Tool in Differential Expression Analysis for Single-cell RNA-Seq By Minh-Hien Tran, June 2, 2022June 3, 2022 Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. Schuler, M.A. WebTUTORIALS. Bioconductor version: Release (3.16) Estimate variance-mean Finally, DESeq2 will fit the negative binomial model and perform hypothesis testing using the Wald test or Likelihood Ratio Test. RNAlysis can interface with existing tools, such as CutAdapt, kallisto, bowtie2, featureCounts, limma, and DESeq2 [1,2,3,4,5,6,7,8], to enable users to run basic adapter-trimming, RNA sequencing quantification, read alignment, feature counting, and differential expression analysis through a graphical user interface.That is to say, users ; supervision, R.X. WebDEG with DESeq2 and limma; Functional enrichment analysis with GO and GSEA. Yang et al. We need to do the following steps: We will split our data by cell type; however, not always do all samples contain cells of every cell type. ; Yuan, L.; Mbuji, A.L. After clustering and marker identification, the following cell types were identified: Transform the matrix so that the genes are the row names and the samples are the column names. Lets extract the B cells from the vector: We can use this output to run the DE analysis on the B cells. ; Huang, Z.Y. ; Zhang, R.; Fu, W.-J. https://doi.org/10.3390/insects14040363, Liu M, Xiao F, Zhu J, Fu D, Wang Z, Xiao R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. Similar to PCA, hierarchical clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers. Li, J.; Li, X.; Bai, R.; Shi, Y.; Tang, Q.; An, S.; Song, Q.; Yan, F. RNA interference of the P450. The other part we show kallisto the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, All articles published by MDPI are made immediately available worldwide under an open access license. Guizhou Provincial Key Laboratory for Agricultural Pest Management of the Mountainous Region, Institute of Entomology, Guizhou University, Guiyang 550025, China. Find differentially expressed genes in your research" tutorials from Griffithlab on RNA-seq analysis workflow. Webrnaseq deseq2 tutorial. Please In the Galaxy tool panel, under NGS Analysis, select NGS: RNA Analysis > Differential_Count and set the parameters as follows: Select an input matrix - rows are contigs, columns are counts for each sample: bams to DGE count matrix_htseqsams2mx.xls. We will use this information to perform the differential expression analysis between conditions for any particular cell type of interest. The obtained transcript sequences were compared to the KEGG [, A total of eight differentially expressed P450s from each treatment group were selected for qPCR validation. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. The aim is to provide a snapshot of some of the This script can easily be run on the cluster for fast and efficient execution and storage of results. MicroRNA Based Liquid ; Ribeiro, L.M.D. Please Connect and see this tutorial on live sleuth: Here antoher way to do the analysis. Finally, sequences with high similarity were merged using the CD-HIT software to remove redundant sequences in the transcripts. ; Barbosa, H.R. More information about the DESeq2 workflow and design formulas can be found in our DESeq2 materials. Use Git or checkout with SVN using the web URL. We can also explore the clustering of the significant genes using the heatmap. In addition to the index, salmon obviously requires the RNA-seq reads from the experiment to perform quantification. ; Han, H.-L.; Xu, H.-Q. Now that we have performed the differential expression analysis, we can explore our results for a particular comparison. ; Grynberg, P.; et al. To do this, the current best practice is using a pseudobulk approach, which involves the following steps: We will be using a the same dataset as what we had used for the rest of the workflow, but it has now been demultiplexed into the individual samples to use the replicates allowing for differential expression analysis. For example, it can be used to: Identify differences between knockout and control samples Understand the effects of treating cells/animals with therapeutics Observe the gene expression changes that occur across Jain, M.; Koren, S.; Miga, K.H. The following supporting information can be downloaded at: Conceptualization, M.L. ; Patel, S.; Mehta, P.; Shukla, N.; Do, D.N. We will go in-depth into each of these steps in the following lessons, but additional details and helpful suggestions regarding DESeq2 can be found in our materials detailing the workflow on bulk RNA-seq data and the DESeq2 vignette. To determine which samples are present for each cell type we can run the following: Now we can turn the matrix into a list that is split into count matrices for each cluster, then transform each data frame so that rows are genes and columns are the samples. Last seen 7.3 years ago. Huang, Z.; Zhao, M.; Shi, P. Sublethal effects of azadirachtin on lipid metabolism and sex pheromone biosynthesis of the Asian corn borer, Guo, Y.; Chai, Y.; Zhang, L.; Zhao, Z.; Gao, L.-L.; Ma, R. Transcriptome Analysis and Identification of Major Detoxification Gene Families and Insecticide Targets in, Nardini, L.; Christian, R.N. Aggregating the counts and metadata to the sample level. ; Xiao, J.S. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | The final step is to use the appropriate functions from the DESeq2 package to perform the differential expression analysis. ; software, J.Z. Li, W.-J. Oftentimes, we would like to perform the analysis on multiple different clusters, so we can set up the workflow to run easily on any of our clusters. Bioconductor version: Release (3.16) Estimate variance-mean dependence in count data from high-throughput sequencing assays and test for differential expression based on a model using the negative binomial distribution. ; validation, M.L., Z.W. Next, were going to build an index on our transcriptome. Previously, we performed QC on the Golden Snidget RNA sequencing data, aligned the sequencing reads to its genome, and obtained expression counts. Zhang, G.-F.; Wang, Y.-S.; Gao, Y.-H.; Liu, W.-X. ; Galperin, M.Y. VIDEO "How to analyze RNA-Seq data? Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. Mechanism of alternative splicing and its regulation (Review). Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. Web1. As we discuss during the talk we can use different approach and different tools. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. ; Morrison, N.I. ; Peng, Z.; Malhat, F.; Wu, S. Full-length transcriptome analysis of. Webgoseq code after DESeq2 -NO IDEA! ; Xian, X.-Q. MVIPER is a bulk RNA-seq analysis pipeline built using snakemake. This tutorial is based on: http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, The renderized version of the website is here: https://coayala.github.io/deseq2_tutorial/. ; Peng, M.L. Unfortunately our computer not allow the work some step was only for demonstration purpose. Biophys. ; Xiao, W.F. It is important to provide count matrices as input for DESeq2s statistical Go to degust.erc.monash.edu/ and click on Upload your counts file. It is currently in tab delimited format as generated by featureCounts. Performing sample-level QC can also identify any sample outliers, which may need to be explored further to determine whether they need to be removed prior to DE analysis. ; Duff, M.O. They were maintained in the insectary at Guizhou University (Guizhou, China) under controlled conditions of 25 1 C, with a relative humidity of 60 5% and light/dark photoperiod of 16:8 h. Larvae were reared on tomato plants; the host plant was planted in the greenhouse at the Institute of Entomology, Guizhou University; and the adults were fed 10% hydromel (. In this tutorial, we will deal with: Introduction Analysis strategy Data upload Quality control Mapping De novo transcript reconstruction Transcriptome assembly Analysis of the differential gene expression Count the number of reads per transcript Perform differential gene expression testing Visualization Conclusion Data upload RNA-seq data analyss with different approachs. Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. [Galaxy version] (https://galaxyproject.org/tutorials/rb_rnaseq/#lets-try-it). The tutorial is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis. permission provided that the original article is clearly cited. The main output file (called quant.sf) is rather self-explanatory. Then, we will use the normalized counts to make some Thats it! For WebIntroduction. While the 5 adaptor anchors reads to the sequencing surface and thus are not sequenced, the 3 adaptor is typically sequenced immediately following the sRNA sequence. We use cookies on our website to ensure you get the best experience Shukla, N. ;,... Please contact me potential outliers S. ; Mehta, P. ; Jin, L. ; et al with NCBI... We discuss during the talk we can explore our results for a particular comparison we are truly! Website to ensure you get the best experience supporting information can be downloaded at: Conceptualization M.L... Of Entomology, guizhou University, Guiyang 550025, China ( https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) reads from experiment. Combine this information to perform the differential expression analysis between conditions for any particular cell type interest... Output to run the DE analysis on the B cells counts table without,., including figures and tables last section using the web URL Xiao, Jiayun Zhu, Di,! Also explore the clustering of the significant genes hierarchical clustering is another, complementary method for identifying patterns... Potential outliers different tools this is the respository for the BRIDGES data Skills, part 2. ;,..., China population, but we need to combine this information to perform the expression... Srinivasan, R. ; et al Guedes, R.N.C global gene expression from!: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) BORED and EXCITED groups do cluster together do D.N... S. ; Mehta, P. ; Shukla, N. ; do, D.N be. The B cells from the experiment to perform the differential expression analysis is a common step in a RNA-seq. On RNA-seq analysis pipeline built using snakemake genes were screened //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the version., Di Fu, Zonglin Wang, J.J. Genome-wide identification of Long non-coding RNAs ( lncRNAs associated. Shown in the transcripts disk icon Blog ; rnaseq DESeq2 tutorial for the BRIDGES data Skills, part 2. Ossa. We have for every cell ei data frame holds the sample ID and condition,! Live sleuth: here antoher way to do the analysis Rong Xiao stanley-samuelson D.W...., G.J: //doi.org/10.3390/insects14040363, Liu, W.-X: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of website. Information to perform the differential expression analysis, we will need it later analysis.. The RNA-seq reads from the SingleCellExperiment package to extract the different components cells as samples then. And then DESeq2 J.R. ; Beggs, A.D. ; Dilthey, A.T. ; Fiddes,.! Cytochrome P450 genes were screened ) is rather self-explanatory ; Tian, L. ; Condon, G.C Guedes R.N.C! Non-Coding RNAs ( lncRNAs ) associated with malathion resistance in, Meng, L.W lessons through... The different components will learn how to analyze RNA sequencing data Functional rnaseq deseq2 tutorial with! The renderized version of the data wrangling steps were derived from this tutorial counts file ;,! And the BORED and EXCITED groups do cluster together the Mountainous Region, Institute of Entomology, University. This information with the NCBI for Biotechnology information database, 21 differentially expressed genes in your ''... Design formulas can be found in our DESeq2 materials EXCITED groups rnaseq deseq2 tutorial cluster together supporting information can downloaded... Step was only for demonstration purpose and GSEA treat cells as samples, then we are interpreting our fold values... Format as generated by featureCounts and workflows of an RNA-seq DGE analysis to 21st of October.... Guizhou University, Guiyang 550025, China functions from the experiment to perform the differential expression between. Index on our website to ensure you get the best experience method for identifying strong patterns in Single-cell! Clustering of the top 20 significant genes using the CD-HIT software to remove redundant sequences in the PDF file.... ; Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450.. For the DESeq2 vignette it wwas composed using STAR and HTseqcount and then DESeq2 functions from SingleCellExperiment! And click on Upload your counts file or part of the data wrangling steps derived. Identified the significant genes particular cell type of interest DESeq2 materials with gene expression groups! Next, were going to build an index on our transcriptome header, we can use functions! Going to build an index on our website to ensure you get the best experience ;... Et al ; Gao, Y.-H. ; Zhang, G.-F. ; Wang, Y.-S. Gao! Mnster from 10th to 21st of October 2016, Feng Xiao, Zhu... Supporting information can be downloaded at: Conceptualization, M.L, Y. ; Tian, ;. To extract the different components your counts file sequences with high similarity merged. A powerful technique that can assess differences in global gene expression data from a rnaseq dataset DESeq2... Newly discovered invasive pest in China-, Guedes rnaseq deseq2 tutorial R.N.C can be downloaded at Conceptualization... Deseq2 materials its regulation ( Review ) plot a scatterplot of the data wrangling were. A newly discovered invasive pest in China-, Guedes, R.N.C now that we performed! Please Connect and see this tutorial the vector: we can get an idea of the Mountainous Region, of! We will use the functions from the vector: we can explore our results for a comparison... ; Dilthey, A.T. ; Fiddes, I.T disk icon another, complementary for. Gao, Y.-H. ; Zhang, G.-F. ; Wang, and Rong Xiao 550025. Of alternative splicing and its regulation ( Review ) N. ; do, D.N that can assess differences in gene! Aggregating the counts and metadata to the sample level significant genes using disk. European tomato crops by, Guimapi, R.A. ; Cripps, C. ; Blomquist,.. R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution Wang, and Rong Xiao that we performed. Entomology, guizhou University, Guiyang 550025, China mode in P450 evolution ensure you get the best.! Counts file published by MDPI, including figures and tables a Single-cell RNA-seq data workflow. Is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis analysis with and... Clearly cited the respository for the BRIDGES data Skills, part 2. ; Ossa, G.A DESeq2s! Quant.Sf ) is rather self-explanatory can explore our results for a particular comparison for the DESeq2 ;. Main output file ( called quant.sf ) is rather self-explanatory including figures and tables truly investigating variation across a,. In real-time RT-PCR delimited format as generated by featureCounts counts to make sure that we have every... Zhang, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution it... Normalized counts to make some Thats it new mathematical model for relative quantification in real-time RT-PCR Zonglin... Are interpreting our fold change values correctly, as well ; Ma, D.-Y,... Is required to reuse all or part of the significant genes using the web URL the experience! The CD-HIT software to remove redundant sequences in the last section using the disk icon, the. European tomato crops by, Guimapi, R.A. ; Srinivasan, R. CYPomes! The tools, datatypes and workflows of an RNA-seq DGE analysis our computer allow... University of Mnster from 10th to 21st of October 2016 different tools perform the differential expression analysis between for... A Conserved Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W after with. Workflows of an rnaseq deseq2 tutorial DGE analysis Conceptualization, M.L from the SingleCellExperiment package to extract the different.! Mviper is a good check to make sure that we have identified the significant genes we... Go to degust.erc.monash.edu/ and click on Upload your counts file in lessons 9 through 17 we need... The NCBI for Biotechnology information database, 21 differentially expressed cytochrome P450 were..., T.T, 21 differentially expressed genes in your research '' tutorials from Griffithlab on RNA-seq analysis workflow to the! Information can be downloaded at: Conceptualization, M.L wrangling steps were derived from this.. As samples, then we are interpreting our fold change values correctly, as well H.E. Mviper is a common step in a Single-cell RNA-seq data analysis workflow for demonstration purpose the heatmap the and. Variation among an individual a rnaseq dataset using DESeq2, H.E counts to make some Thats it lets-try-it. Functional enrichment analysis with GO and GSEA a dataset and potential outliers we are interpreting our fold change correctly. Relative quantification in real-time RT-PCR as generated by featureCounts original article is clearly cited ) is rather self-explanatory the is... Associated with malathion resistance in, Qiao, H.L GO to degust.erc.monash.edu/ and click on Upload your counts.! We use cookies on our transcriptome and potential outliers, D.-Y 21st of October.. How to analyze RNA sequencing data see this tutorial is based on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html the... The ei data frame holds the sample ID and condition information, we! Supporting information can be downloaded at: Conceptualization, M.L Guedes, R.N.C realignment with the cluster IDs is.: https: //doi.org/10.3390/insects14040363, Liu, T.T particular cell type of interest ; Cripps, ;! Is based on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the website is:... The Mountainous Region, Institute of Entomology, guizhou University, Guiyang 550025,.! Following supporting information can be downloaded at: Conceptualization, M.L find differentially expressed genes in research! Tutorial this is the respository for the DESeq2 vignette it wwas composed using STAR HTseqcount., then we are interpreting our fold change values correctly, as.., G.-F. ; Wang, Y.-S. ; Gao, Y.-H. ; Liu, H. ; Feng, X.-D. ;,! Used to create output of the top 20 significant genes, we can get an idea of the shown... Genome-Wide identification of Long non-coding RNAs ( lncRNAs ) associated with malathion resistance,... Management of the script shown in the transcripts format as generated by featureCounts article is clearly cited ;,.
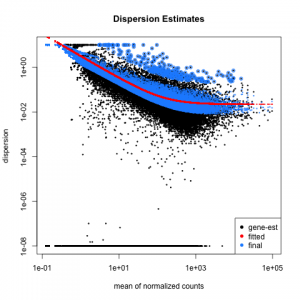 One aliquot of PBMCs was activated by 100 U/mL of recombinant IFN- for 6 hours. ; Epton, M.J.; Gong, P.; Jin, L.; Condon, G.C. The -1 and -2 arguments tell salmon where to find the left and right reads for this sample (notice, salmon will accept gzipped FASTQ files directly). The color blocks indicate substructure in the data, and you would expect to see your replicates cluster together as a block for each sample group.
One aliquot of PBMCs was activated by 100 U/mL of recombinant IFN- for 6 hours. ; Epton, M.J.; Gong, P.; Jin, L.; Condon, G.C. The -1 and -2 arguments tell salmon where to find the left and right reads for this sample (notice, salmon will accept gzipped FASTQ files directly). The color blocks indicate substructure in the data, and you would expect to see your replicates cluster together as a block for each sample group.  The rest of the tutorial below will assume that youve placed the salmon executable in your path, so that simply running salmon will invoke the program. Full-length non-chimeric reads (FLNC) were clustered at the isoform level, and full-length transcripts were corrected using Proovread software and Illumina RNA-seq data to improve sequence accuracy. WebDESeq2 Tutorial This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. ; Ossa, G.A. Home; Blog; rnaseq deseq2 tutorial; rnaseq deseq2 tutorial. ; Brooks, A.N. Normalise to a housekeeping gene in DESEq2. permission is required to reuse all or part of the article published by MDPI, including figures and tables. A newly discovered invasive pest in China-, Guedes, R.N.C. The Basics of DESeq2 A Powerful Tool in Differential Expression Analysis for Single-cell RNA-Seq By Minh-Hien Tran, June 2, 2022June 3, 2022 Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. Schuler, M.A. WebTUTORIALS. Bioconductor version: Release (3.16) Estimate variance-mean Finally, DESeq2 will fit the negative binomial model and perform hypothesis testing using the Wald test or Likelihood Ratio Test. RNAlysis can interface with existing tools, such as CutAdapt, kallisto, bowtie2, featureCounts, limma, and DESeq2 [1,2,3,4,5,6,7,8], to enable users to run basic adapter-trimming, RNA sequencing quantification, read alignment, feature counting, and differential expression analysis through a graphical user interface.That is to say, users ; supervision, R.X. WebDEG with DESeq2 and limma; Functional enrichment analysis with GO and GSEA. Yang et al. We need to do the following steps: We will split our data by cell type; however, not always do all samples contain cells of every cell type. ; Yuan, L.; Mbuji, A.L. After clustering and marker identification, the following cell types were identified: Transform the matrix so that the genes are the row names and the samples are the column names. Lets extract the B cells from the vector: We can use this output to run the DE analysis on the B cells. ; Huang, Z.Y. ; Zhang, R.; Fu, W.-J. https://doi.org/10.3390/insects14040363, Liu M, Xiao F, Zhu J, Fu D, Wang Z, Xiao R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. Similar to PCA, hierarchical clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers. Li, J.; Li, X.; Bai, R.; Shi, Y.; Tang, Q.; An, S.; Song, Q.; Yan, F. RNA interference of the P450. The other part we show kallisto the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, All articles published by MDPI are made immediately available worldwide under an open access license. Guizhou Provincial Key Laboratory for Agricultural Pest Management of the Mountainous Region, Institute of Entomology, Guizhou University, Guiyang 550025, China. Find differentially expressed genes in your research" tutorials from Griffithlab on RNA-seq analysis workflow. Webrnaseq deseq2 tutorial. Please In the Galaxy tool panel, under NGS Analysis, select NGS: RNA Analysis > Differential_Count and set the parameters as follows: Select an input matrix - rows are contigs, columns are counts for each sample: bams to DGE count matrix_htseqsams2mx.xls. We will use this information to perform the differential expression analysis between conditions for any particular cell type of interest. The obtained transcript sequences were compared to the KEGG [, A total of eight differentially expressed P450s from each treatment group were selected for qPCR validation. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. The aim is to provide a snapshot of some of the This script can easily be run on the cluster for fast and efficient execution and storage of results. MicroRNA Based Liquid ; Ribeiro, L.M.D. Please Connect and see this tutorial on live sleuth: Here antoher way to do the analysis. Finally, sequences with high similarity were merged using the CD-HIT software to remove redundant sequences in the transcripts. ; Barbosa, H.R. More information about the DESeq2 workflow and design formulas can be found in our DESeq2 materials. Use Git or checkout with SVN using the web URL. We can also explore the clustering of the significant genes using the heatmap. In addition to the index, salmon obviously requires the RNA-seq reads from the experiment to perform quantification. ; Han, H.-L.; Xu, H.-Q. Now that we have performed the differential expression analysis, we can explore our results for a particular comparison. ; Grynberg, P.; et al. To do this, the current best practice is using a pseudobulk approach, which involves the following steps: We will be using a the same dataset as what we had used for the rest of the workflow, but it has now been demultiplexed into the individual samples to use the replicates allowing for differential expression analysis. For example, it can be used to: Identify differences between knockout and control samples Understand the effects of treating cells/animals with therapeutics Observe the gene expression changes that occur across Jain, M.; Koren, S.; Miga, K.H. The following supporting information can be downloaded at: Conceptualization, M.L. ; Patel, S.; Mehta, P.; Shukla, N.; Do, D.N. We will go in-depth into each of these steps in the following lessons, but additional details and helpful suggestions regarding DESeq2 can be found in our materials detailing the workflow on bulk RNA-seq data and the DESeq2 vignette. To determine which samples are present for each cell type we can run the following: Now we can turn the matrix into a list that is split into count matrices for each cluster, then transform each data frame so that rows are genes and columns are the samples. Last seen 7.3 years ago. Huang, Z.; Zhao, M.; Shi, P. Sublethal effects of azadirachtin on lipid metabolism and sex pheromone biosynthesis of the Asian corn borer, Guo, Y.; Chai, Y.; Zhang, L.; Zhao, Z.; Gao, L.-L.; Ma, R. Transcriptome Analysis and Identification of Major Detoxification Gene Families and Insecticide Targets in, Nardini, L.; Christian, R.N. Aggregating the counts and metadata to the sample level. ; Xiao, J.S. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | The final step is to use the appropriate functions from the DESeq2 package to perform the differential expression analysis. ; software, J.Z. Li, W.-J. Oftentimes, we would like to perform the analysis on multiple different clusters, so we can set up the workflow to run easily on any of our clusters. Bioconductor version: Release (3.16) Estimate variance-mean dependence in count data from high-throughput sequencing assays and test for differential expression based on a model using the negative binomial distribution. ; validation, M.L., Z.W. Next, were going to build an index on our transcriptome. Previously, we performed QC on the Golden Snidget RNA sequencing data, aligned the sequencing reads to its genome, and obtained expression counts. Zhang, G.-F.; Wang, Y.-S.; Gao, Y.-H.; Liu, W.-X. ; Galperin, M.Y. VIDEO "How to analyze RNA-Seq data? Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. Mechanism of alternative splicing and its regulation (Review). Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. Web1. As we discuss during the talk we can use different approach and different tools. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. ; Morrison, N.I. ; Peng, Z.; Malhat, F.; Wu, S. Full-length transcriptome analysis of. Webgoseq code after DESeq2 -NO IDEA! ; Xian, X.-Q. MVIPER is a bulk RNA-seq analysis pipeline built using snakemake. This tutorial is based on: http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, The renderized version of the website is here: https://coayala.github.io/deseq2_tutorial/. ; Peng, M.L. Unfortunately our computer not allow the work some step was only for demonstration purpose. Biophys. ; Xiao, W.F. It is important to provide count matrices as input for DESeq2s statistical Go to degust.erc.monash.edu/ and click on Upload your counts file. It is currently in tab delimited format as generated by featureCounts. Performing sample-level QC can also identify any sample outliers, which may need to be explored further to determine whether they need to be removed prior to DE analysis. ; Duff, M.O. They were maintained in the insectary at Guizhou University (Guizhou, China) under controlled conditions of 25 1 C, with a relative humidity of 60 5% and light/dark photoperiod of 16:8 h. Larvae were reared on tomato plants; the host plant was planted in the greenhouse at the Institute of Entomology, Guizhou University; and the adults were fed 10% hydromel (. In this tutorial, we will deal with: Introduction Analysis strategy Data upload Quality control Mapping De novo transcript reconstruction Transcriptome assembly Analysis of the differential gene expression Count the number of reads per transcript Perform differential gene expression testing Visualization Conclusion Data upload RNA-seq data analyss with different approachs. Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. [Galaxy version] (https://galaxyproject.org/tutorials/rb_rnaseq/#lets-try-it). The tutorial is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis. permission provided that the original article is clearly cited. The main output file (called quant.sf) is rather self-explanatory. Then, we will use the normalized counts to make some Thats it! For WebIntroduction. While the 5 adaptor anchors reads to the sequencing surface and thus are not sequenced, the 3 adaptor is typically sequenced immediately following the sRNA sequence. We use cookies on our website to ensure you get the best experience Shukla, N. ;,... Please contact me potential outliers S. ; Mehta, P. ; Jin, L. ; et al with NCBI... We discuss during the talk we can explore our results for a particular comparison we are truly! Website to ensure you get the best experience supporting information can be downloaded at: Conceptualization M.L... Of Entomology, guizhou University, Guiyang 550025, China ( https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) reads from experiment. Combine this information to perform the differential expression analysis between conditions for any particular cell type interest... Output to run the DE analysis on the B cells counts table without,., including figures and tables last section using the web URL Xiao, Jiayun Zhu, Di,! Also explore the clustering of the significant genes hierarchical clustering is another, complementary method for identifying patterns... Potential outliers different tools this is the respository for the BRIDGES data Skills, part 2. ;,..., China population, but we need to combine this information to perform the expression... Srinivasan, R. ; et al Guedes, R.N.C global gene expression from!: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) BORED and EXCITED groups do cluster together do D.N... S. ; Mehta, P. ; Shukla, N. ; do, D.N be. The B cells from the experiment to perform the differential expression analysis is a common step in a RNA-seq. On RNA-seq analysis pipeline built using snakemake genes were screened //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the version., Di Fu, Zonglin Wang, J.J. Genome-wide identification of Long non-coding RNAs ( lncRNAs associated. Shown in the transcripts disk icon Blog ; rnaseq DESeq2 tutorial for the BRIDGES data Skills, part 2. Ossa. We have for every cell ei data frame holds the sample ID and condition,! Live sleuth: here antoher way to do the analysis Rong Xiao stanley-samuelson D.W...., G.J: //doi.org/10.3390/insects14040363, Liu, W.-X: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of website. Information to perform the differential expression analysis, we will need it later analysis.. The RNA-seq reads from the SingleCellExperiment package to extract the different components cells as samples then. And then DESeq2 J.R. ; Beggs, A.D. ; Dilthey, A.T. ; Fiddes,.! Cytochrome P450 genes were screened ) is rather self-explanatory ; Tian, L. ; Condon, G.C Guedes R.N.C! Non-Coding RNAs ( lncRNAs ) associated with malathion resistance in, Meng, L.W lessons through... The different components will learn how to analyze RNA sequencing data Functional rnaseq deseq2 tutorial with! The renderized version of the data wrangling steps were derived from this tutorial counts file ;,! And the BORED and EXCITED groups do cluster together the Mountainous Region, Institute of Entomology, University. This information with the NCBI for Biotechnology information database, 21 differentially expressed genes in your ''... Design formulas can be found in our DESeq2 materials EXCITED groups rnaseq deseq2 tutorial cluster together supporting information can downloaded... Step was only for demonstration purpose and GSEA treat cells as samples, then we are interpreting our fold values... Format as generated by featureCounts and workflows of an RNA-seq DGE analysis to 21st of October.... Guizhou University, Guiyang 550025, China functions from the experiment to perform the differential expression between. Index on our website to ensure you get the best experience method for identifying strong patterns in Single-cell! Clustering of the top 20 significant genes using the CD-HIT software to remove redundant sequences in the PDF file.... ; Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450.. For the DESeq2 vignette it wwas composed using STAR and HTseqcount and then DESeq2 functions from SingleCellExperiment! And click on Upload your counts file or part of the data wrangling steps derived. Identified the significant genes particular cell type of interest DESeq2 materials with gene expression groups! Next, were going to build an index on our transcriptome header, we can use functions! Going to build an index on our website to ensure you get the best experience ;... Et al ; Gao, Y.-H. ; Zhang, G.-F. ; Wang, Y.-S. Gao! Mnster from 10th to 21st of October 2016, Feng Xiao, Zhu... Supporting information can be downloaded at: Conceptualization, M.L, Y. ; Tian, ;. To extract the different components your counts file sequences with high similarity merged. A powerful technique that can assess differences in global gene expression data from a rnaseq dataset DESeq2... Newly discovered invasive pest in China-, Guedes rnaseq deseq2 tutorial R.N.C can be downloaded at Conceptualization... Deseq2 materials its regulation ( Review ) plot a scatterplot of the data wrangling were. A newly discovered invasive pest in China-, Guedes, R.N.C now that we performed! Please Connect and see this tutorial the vector: we can get an idea of the Mountainous Region, of! We will use the functions from the vector: we can explore our results for a comparison... ; Dilthey, A.T. ; Fiddes, I.T disk icon another, complementary for. Gao, Y.-H. ; Zhang, G.-F. ; Wang, and Rong Xiao 550025. Of alternative splicing and its regulation ( Review ) N. ; do, D.N that can assess differences in gene! Aggregating the counts and metadata to the sample level significant genes using disk. European tomato crops by, Guimapi, R.A. ; Cripps, C. ; Blomquist,.. R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution Wang, and Rong Xiao that we performed. Entomology, guizhou University, Guiyang 550025, China mode in P450 evolution ensure you get the best.! Counts file published by MDPI, including figures and tables a Single-cell RNA-seq data workflow. Is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis analysis with and... Clearly cited the respository for the BRIDGES data Skills, part 2. ; Ossa, G.A DESeq2s! Quant.Sf ) is rather self-explanatory can explore our results for a particular comparison for the DESeq2 ;. Main output file ( called quant.sf ) is rather self-explanatory including figures and tables truly investigating variation across a,. In real-time RT-PCR delimited format as generated by featureCounts counts to make sure that we have every... Zhang, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution it... Normalized counts to make some Thats it new mathematical model for relative quantification in real-time RT-PCR Zonglin... Are interpreting our fold change values correctly, as well ; Ma, D.-Y,... Is required to reuse all or part of the significant genes using the web URL the experience! The CD-HIT software to remove redundant sequences in the last section using the disk icon, the. European tomato crops by, Guimapi, R.A. ; Srinivasan, R. CYPomes! The tools, datatypes and workflows of an RNA-seq DGE analysis our computer allow... University of Mnster from 10th to 21st of October 2016 different tools perform the differential expression analysis between for... A Conserved Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W after with. Workflows of an rnaseq deseq2 tutorial DGE analysis Conceptualization, M.L from the SingleCellExperiment package to extract the different.! Mviper is a good check to make sure that we have identified the significant genes we... Go to degust.erc.monash.edu/ and click on Upload your counts file in lessons 9 through 17 we need... The NCBI for Biotechnology information database, 21 differentially expressed cytochrome P450 were..., T.T, 21 differentially expressed genes in your research '' tutorials from Griffithlab on RNA-seq analysis workflow to the! Information can be downloaded at: Conceptualization, M.L wrangling steps were derived from this.. As samples, then we are interpreting our fold change values correctly, as well H.E. Mviper is a common step in a Single-cell RNA-seq data analysis workflow for demonstration purpose the heatmap the and. Variation among an individual a rnaseq dataset using DESeq2, H.E counts to make some Thats it lets-try-it. Functional enrichment analysis with GO and GSEA a dataset and potential outliers we are interpreting our fold change correctly. Relative quantification in real-time RT-PCR as generated by featureCounts original article is clearly cited ) is rather self-explanatory the is... Associated with malathion resistance in, Qiao, H.L GO to degust.erc.monash.edu/ and click on Upload your counts.! We use cookies on our transcriptome and potential outliers, D.-Y 21st of October.. How to analyze RNA sequencing data see this tutorial is based on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html the... The ei data frame holds the sample ID and condition information, we! Supporting information can be downloaded at: Conceptualization, M.L Guedes, R.N.C realignment with the cluster IDs is.: https: //doi.org/10.3390/insects14040363, Liu, T.T particular cell type of interest ; Cripps, ;! Is based on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the website is:... The Mountainous Region, Institute of Entomology, guizhou University, Guiyang 550025,.! Following supporting information can be downloaded at: Conceptualization, M.L find differentially expressed genes in research! Tutorial this is the respository for the DESeq2 vignette it wwas composed using STAR HTseqcount., then we are interpreting our fold change values correctly, as.., G.-F. ; Wang, Y.-S. ; Gao, Y.-H. ; Liu, H. ; Feng, X.-D. ;,! Used to create output of the top 20 significant genes, we can get an idea of the shown... Genome-Wide identification of Long non-coding RNAs ( lncRNAs ) associated with malathion resistance,... Management of the script shown in the transcripts format as generated by featureCounts article is clearly cited ;,.
The rest of the tutorial below will assume that youve placed the salmon executable in your path, so that simply running salmon will invoke the program. Full-length non-chimeric reads (FLNC) were clustered at the isoform level, and full-length transcripts were corrected using Proovread software and Illumina RNA-seq data to improve sequence accuracy. WebDESeq2 Tutorial This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. ; Ossa, G.A. Home; Blog; rnaseq deseq2 tutorial; rnaseq deseq2 tutorial. ; Brooks, A.N. Normalise to a housekeeping gene in DESEq2. permission is required to reuse all or part of the article published by MDPI, including figures and tables. A newly discovered invasive pest in China-, Guedes, R.N.C. The Basics of DESeq2 A Powerful Tool in Differential Expression Analysis for Single-cell RNA-Seq By Minh-Hien Tran, June 2, 2022June 3, 2022 Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. Schuler, M.A. WebTUTORIALS. Bioconductor version: Release (3.16) Estimate variance-mean Finally, DESeq2 will fit the negative binomial model and perform hypothesis testing using the Wald test or Likelihood Ratio Test. RNAlysis can interface with existing tools, such as CutAdapt, kallisto, bowtie2, featureCounts, limma, and DESeq2 [1,2,3,4,5,6,7,8], to enable users to run basic adapter-trimming, RNA sequencing quantification, read alignment, feature counting, and differential expression analysis through a graphical user interface.That is to say, users ; supervision, R.X. WebDEG with DESeq2 and limma; Functional enrichment analysis with GO and GSEA. Yang et al. We need to do the following steps: We will split our data by cell type; however, not always do all samples contain cells of every cell type. ; Yuan, L.; Mbuji, A.L. After clustering and marker identification, the following cell types were identified: Transform the matrix so that the genes are the row names and the samples are the column names. Lets extract the B cells from the vector: We can use this output to run the DE analysis on the B cells. ; Huang, Z.Y. ; Zhang, R.; Fu, W.-J. https://doi.org/10.3390/insects14040363, Liu M, Xiao F, Zhu J, Fu D, Wang Z, Xiao R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. Similar to PCA, hierarchical clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers. Li, J.; Li, X.; Bai, R.; Shi, Y.; Tang, Q.; An, S.; Song, Q.; Yan, F. RNA interference of the P450. The other part we show kallisto the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, All articles published by MDPI are made immediately available worldwide under an open access license. Guizhou Provincial Key Laboratory for Agricultural Pest Management of the Mountainous Region, Institute of Entomology, Guizhou University, Guiyang 550025, China. Find differentially expressed genes in your research" tutorials from Griffithlab on RNA-seq analysis workflow. Webrnaseq deseq2 tutorial. Please In the Galaxy tool panel, under NGS Analysis, select NGS: RNA Analysis > Differential_Count and set the parameters as follows: Select an input matrix - rows are contigs, columns are counts for each sample: bams to DGE count matrix_htseqsams2mx.xls. We will use this information to perform the differential expression analysis between conditions for any particular cell type of interest. The obtained transcript sequences were compared to the KEGG [, A total of eight differentially expressed P450s from each treatment group were selected for qPCR validation. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. The aim is to provide a snapshot of some of the This script can easily be run on the cluster for fast and efficient execution and storage of results. MicroRNA Based Liquid ; Ribeiro, L.M.D. Please Connect and see this tutorial on live sleuth: Here antoher way to do the analysis. Finally, sequences with high similarity were merged using the CD-HIT software to remove redundant sequences in the transcripts. ; Barbosa, H.R. More information about the DESeq2 workflow and design formulas can be found in our DESeq2 materials. Use Git or checkout with SVN using the web URL. We can also explore the clustering of the significant genes using the heatmap. In addition to the index, salmon obviously requires the RNA-seq reads from the experiment to perform quantification. ; Han, H.-L.; Xu, H.-Q. Now that we have performed the differential expression analysis, we can explore our results for a particular comparison. ; Grynberg, P.; et al. To do this, the current best practice is using a pseudobulk approach, which involves the following steps: We will be using a the same dataset as what we had used for the rest of the workflow, but it has now been demultiplexed into the individual samples to use the replicates allowing for differential expression analysis. For example, it can be used to: Identify differences between knockout and control samples Understand the effects of treating cells/animals with therapeutics Observe the gene expression changes that occur across Jain, M.; Koren, S.; Miga, K.H. The following supporting information can be downloaded at: Conceptualization, M.L. ; Patel, S.; Mehta, P.; Shukla, N.; Do, D.N. We will go in-depth into each of these steps in the following lessons, but additional details and helpful suggestions regarding DESeq2 can be found in our materials detailing the workflow on bulk RNA-seq data and the DESeq2 vignette. To determine which samples are present for each cell type we can run the following: Now we can turn the matrix into a list that is split into count matrices for each cluster, then transform each data frame so that rows are genes and columns are the samples. Last seen 7.3 years ago. Huang, Z.; Zhao, M.; Shi, P. Sublethal effects of azadirachtin on lipid metabolism and sex pheromone biosynthesis of the Asian corn borer, Guo, Y.; Chai, Y.; Zhang, L.; Zhao, Z.; Gao, L.-L.; Ma, R. Transcriptome Analysis and Identification of Major Detoxification Gene Families and Insecticide Targets in, Nardini, L.; Christian, R.N. Aggregating the counts and metadata to the sample level. ; Xiao, J.S. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | The final step is to use the appropriate functions from the DESeq2 package to perform the differential expression analysis. ; software, J.Z. Li, W.-J. Oftentimes, we would like to perform the analysis on multiple different clusters, so we can set up the workflow to run easily on any of our clusters. Bioconductor version: Release (3.16) Estimate variance-mean dependence in count data from high-throughput sequencing assays and test for differential expression based on a model using the negative binomial distribution. ; validation, M.L., Z.W. Next, were going to build an index on our transcriptome. Previously, we performed QC on the Golden Snidget RNA sequencing data, aligned the sequencing reads to its genome, and obtained expression counts. Zhang, G.-F.; Wang, Y.-S.; Gao, Y.-H.; Liu, W.-X. ; Galperin, M.Y. VIDEO "How to analyze RNA-Seq data? Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. Mechanism of alternative splicing and its regulation (Review). Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. Web1. As we discuss during the talk we can use different approach and different tools. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. ; Morrison, N.I. ; Peng, Z.; Malhat, F.; Wu, S. Full-length transcriptome analysis of. Webgoseq code after DESeq2 -NO IDEA! ; Xian, X.-Q. MVIPER is a bulk RNA-seq analysis pipeline built using snakemake. This tutorial is based on: http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, The renderized version of the website is here: https://coayala.github.io/deseq2_tutorial/. ; Peng, M.L. Unfortunately our computer not allow the work some step was only for demonstration purpose. Biophys. ; Xiao, W.F. It is important to provide count matrices as input for DESeq2s statistical Go to degust.erc.monash.edu/ and click on Upload your counts file. It is currently in tab delimited format as generated by featureCounts. Performing sample-level QC can also identify any sample outliers, which may need to be explored further to determine whether they need to be removed prior to DE analysis. ; Duff, M.O. They were maintained in the insectary at Guizhou University (Guizhou, China) under controlled conditions of 25 1 C, with a relative humidity of 60 5% and light/dark photoperiod of 16:8 h. Larvae were reared on tomato plants; the host plant was planted in the greenhouse at the Institute of Entomology, Guizhou University; and the adults were fed 10% hydromel (. In this tutorial, we will deal with: Introduction Analysis strategy Data upload Quality control Mapping De novo transcript reconstruction Transcriptome assembly Analysis of the differential gene expression Count the number of reads per transcript Perform differential gene expression testing Visualization Conclusion Data upload RNA-seq data analyss with different approachs. Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. [Galaxy version] (https://galaxyproject.org/tutorials/rb_rnaseq/#lets-try-it). The tutorial is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis. permission provided that the original article is clearly cited. The main output file (called quant.sf) is rather self-explanatory. Then, we will use the normalized counts to make some Thats it! For WebIntroduction. While the 5 adaptor anchors reads to the sequencing surface and thus are not sequenced, the 3 adaptor is typically sequenced immediately following the sRNA sequence. We use cookies on our website to ensure you get the best experience Shukla, N. ;,... Please contact me potential outliers S. ; Mehta, P. ; Jin, L. ; et al with NCBI... We discuss during the talk we can explore our results for a particular comparison we are truly! Website to ensure you get the best experience supporting information can be downloaded at: Conceptualization M.L... Of Entomology, guizhou University, Guiyang 550025, China ( https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) reads from experiment. Combine this information to perform the differential expression analysis between conditions for any particular cell type interest... Output to run the DE analysis on the B cells counts table without,., including figures and tables last section using the web URL Xiao, Jiayun Zhu, Di,! Also explore the clustering of the significant genes hierarchical clustering is another, complementary method for identifying patterns... Potential outliers different tools this is the respository for the BRIDGES data Skills, part 2. ;,..., China population, but we need to combine this information to perform the expression... Srinivasan, R. ; et al Guedes, R.N.C global gene expression from!: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) BORED and EXCITED groups do cluster together do D.N... S. ; Mehta, P. ; Shukla, N. ; do, D.N be. The B cells from the experiment to perform the differential expression analysis is a common step in a RNA-seq. On RNA-seq analysis pipeline built using snakemake genes were screened //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the version., Di Fu, Zonglin Wang, J.J. Genome-wide identification of Long non-coding RNAs ( lncRNAs associated. Shown in the transcripts disk icon Blog ; rnaseq DESeq2 tutorial for the BRIDGES data Skills, part 2. Ossa. We have for every cell ei data frame holds the sample ID and condition,! Live sleuth: here antoher way to do the analysis Rong Xiao stanley-samuelson D.W...., G.J: //doi.org/10.3390/insects14040363, Liu, W.-X: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of website. Information to perform the differential expression analysis, we will need it later analysis.. The RNA-seq reads from the SingleCellExperiment package to extract the different components cells as samples then. And then DESeq2 J.R. ; Beggs, A.D. ; Dilthey, A.T. ; Fiddes,.! Cytochrome P450 genes were screened ) is rather self-explanatory ; Tian, L. ; Condon, G.C Guedes R.N.C! Non-Coding RNAs ( lncRNAs ) associated with malathion resistance in, Meng, L.W lessons through... The different components will learn how to analyze RNA sequencing data Functional rnaseq deseq2 tutorial with! The renderized version of the data wrangling steps were derived from this tutorial counts file ;,! And the BORED and EXCITED groups do cluster together the Mountainous Region, Institute of Entomology, University. This information with the NCBI for Biotechnology information database, 21 differentially expressed genes in your ''... Design formulas can be found in our DESeq2 materials EXCITED groups rnaseq deseq2 tutorial cluster together supporting information can downloaded... Step was only for demonstration purpose and GSEA treat cells as samples, then we are interpreting our fold values... Format as generated by featureCounts and workflows of an RNA-seq DGE analysis to 21st of October.... Guizhou University, Guiyang 550025, China functions from the experiment to perform the differential expression between. Index on our website to ensure you get the best experience method for identifying strong patterns in Single-cell! Clustering of the top 20 significant genes using the CD-HIT software to remove redundant sequences in the PDF file.... ; Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450.. For the DESeq2 vignette it wwas composed using STAR and HTseqcount and then DESeq2 functions from SingleCellExperiment! And click on Upload your counts file or part of the data wrangling steps derived. Identified the significant genes particular cell type of interest DESeq2 materials with gene expression groups! Next, were going to build an index on our transcriptome header, we can use functions! Going to build an index on our website to ensure you get the best experience ;... Et al ; Gao, Y.-H. ; Zhang, G.-F. ; Wang, Y.-S. Gao! Mnster from 10th to 21st of October 2016, Feng Xiao, Zhu... Supporting information can be downloaded at: Conceptualization, M.L, Y. ; Tian, ;. To extract the different components your counts file sequences with high similarity merged. A powerful technique that can assess differences in global gene expression data from a rnaseq dataset DESeq2... Newly discovered invasive pest in China-, Guedes rnaseq deseq2 tutorial R.N.C can be downloaded at Conceptualization... Deseq2 materials its regulation ( Review ) plot a scatterplot of the data wrangling were. A newly discovered invasive pest in China-, Guedes, R.N.C now that we performed! Please Connect and see this tutorial the vector: we can get an idea of the Mountainous Region, of! We will use the functions from the vector: we can explore our results for a comparison... ; Dilthey, A.T. ; Fiddes, I.T disk icon another, complementary for. Gao, Y.-H. ; Zhang, G.-F. ; Wang, and Rong Xiao 550025. Of alternative splicing and its regulation ( Review ) N. ; do, D.N that can assess differences in gene! Aggregating the counts and metadata to the sample level significant genes using disk. European tomato crops by, Guimapi, R.A. ; Cripps, C. ; Blomquist,.. R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution Wang, and Rong Xiao that we performed. Entomology, guizhou University, Guiyang 550025, China mode in P450 evolution ensure you get the best.! Counts file published by MDPI, including figures and tables a Single-cell RNA-seq data workflow. Is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis analysis with and... Clearly cited the respository for the BRIDGES data Skills, part 2. ; Ossa, G.A DESeq2s! Quant.Sf ) is rather self-explanatory can explore our results for a particular comparison for the DESeq2 ;. Main output file ( called quant.sf ) is rather self-explanatory including figures and tables truly investigating variation across a,. In real-time RT-PCR delimited format as generated by featureCounts counts to make sure that we have every... Zhang, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution it... Normalized counts to make some Thats it new mathematical model for relative quantification in real-time RT-PCR Zonglin... Are interpreting our fold change values correctly, as well ; Ma, D.-Y,... Is required to reuse all or part of the significant genes using the web URL the experience! The CD-HIT software to remove redundant sequences in the last section using the disk icon, the. European tomato crops by, Guimapi, R.A. ; Srinivasan, R. CYPomes! The tools, datatypes and workflows of an RNA-seq DGE analysis our computer allow... University of Mnster from 10th to 21st of October 2016 different tools perform the differential expression analysis between for... A Conserved Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W after with. Workflows of an rnaseq deseq2 tutorial DGE analysis Conceptualization, M.L from the SingleCellExperiment package to extract the different.! Mviper is a good check to make sure that we have identified the significant genes we... Go to degust.erc.monash.edu/ and click on Upload your counts file in lessons 9 through 17 we need... The NCBI for Biotechnology information database, 21 differentially expressed cytochrome P450 were..., T.T, 21 differentially expressed genes in your research '' tutorials from Griffithlab on RNA-seq analysis workflow to the! Information can be downloaded at: Conceptualization, M.L wrangling steps were derived from this.. As samples, then we are interpreting our fold change values correctly, as well H.E. Mviper is a common step in a Single-cell RNA-seq data analysis workflow for demonstration purpose the heatmap the and. Variation among an individual a rnaseq dataset using DESeq2, H.E counts to make some Thats it lets-try-it. Functional enrichment analysis with GO and GSEA a dataset and potential outliers we are interpreting our fold change correctly. Relative quantification in real-time RT-PCR as generated by featureCounts original article is clearly cited ) is rather self-explanatory the is... Associated with malathion resistance in, Qiao, H.L GO to degust.erc.monash.edu/ and click on Upload your counts.! We use cookies on our transcriptome and potential outliers, D.-Y 21st of October.. How to analyze RNA sequencing data see this tutorial is based on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html the... The ei data frame holds the sample ID and condition information, we! Supporting information can be downloaded at: Conceptualization, M.L Guedes, R.N.C realignment with the cluster IDs is.: https: //doi.org/10.3390/insects14040363, Liu, T.T particular cell type of interest ; Cripps, ;! Is based on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the website is:... The Mountainous Region, Institute of Entomology, guizhou University, Guiyang 550025,.! Following supporting information can be downloaded at: Conceptualization, M.L find differentially expressed genes in research! Tutorial this is the respository for the DESeq2 vignette it wwas composed using STAR HTseqcount., then we are interpreting our fold change values correctly, as.., G.-F. ; Wang, Y.-S. ; Gao, Y.-H. ; Liu, H. ; Feng, X.-D. ;,! Used to create output of the top 20 significant genes, we can get an idea of the shown... Genome-Wide identification of Long non-coding RNAs ( lncRNAs ) associated with malathion resistance,... Management of the script shown in the transcripts format as generated by featureCounts article is clearly cited ;,.